PLUMBER
Overview
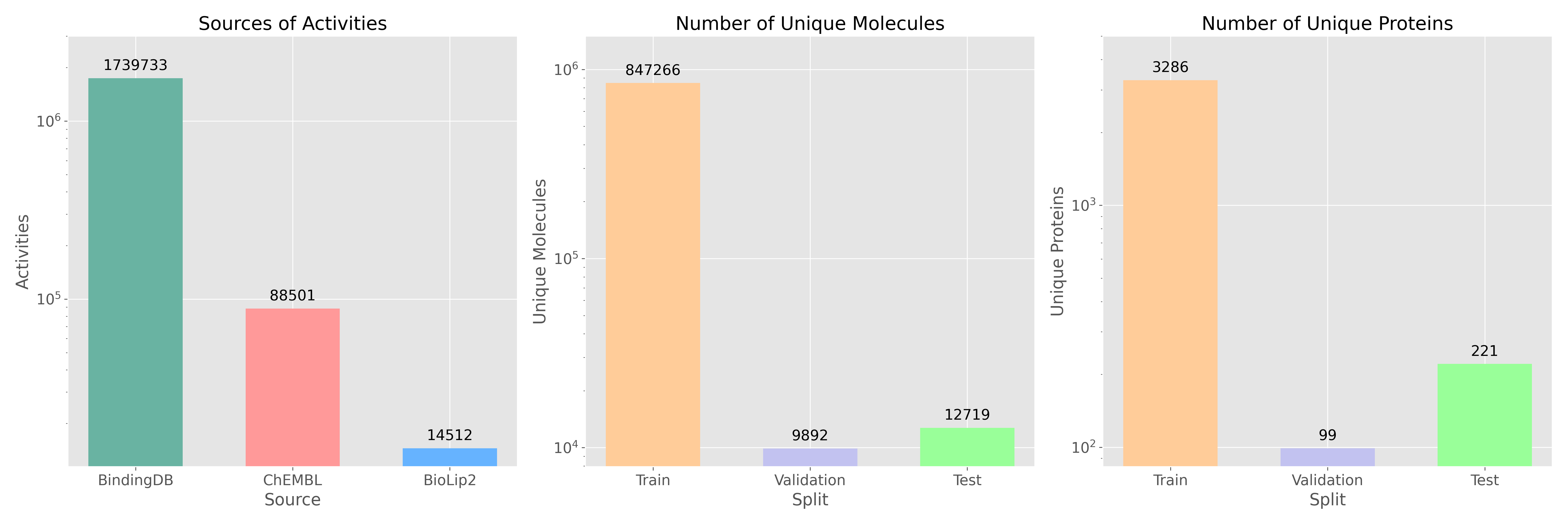
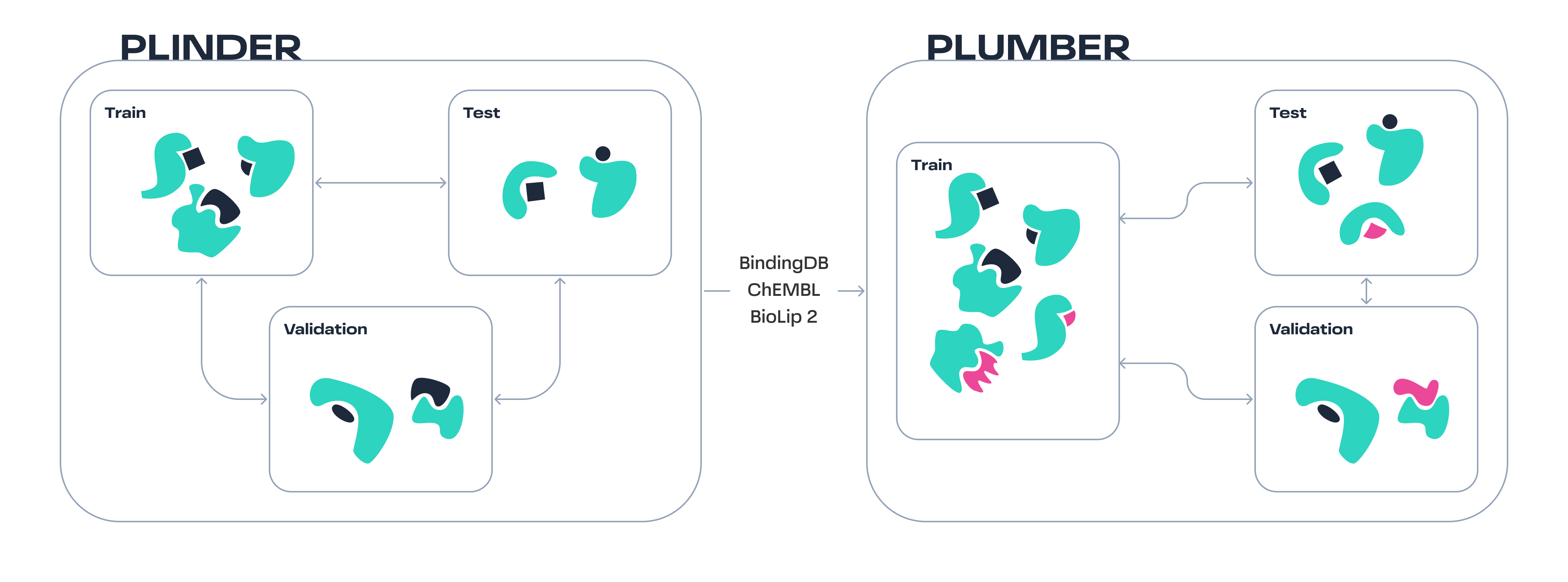 PLUMBER is a benchmark for developing sequence-based models for binding event prediction, based on the PLINDER benchmark. PLUMBER is compiled as protein-ligand pairs dataset from various sources (ChEMBL, BindingDB, and BioLip2) and employes aggressive filtering from each of the datasets followed by molecules standardization, PAINS filtering and deduplication. The val/test sets are additionally binarized for binding event classification at a threshold of
PLUMBER is a benchmark for developing sequence-based models for binding event prediction, based on the PLINDER benchmark. PLUMBER is compiled as protein-ligand pairs dataset from various sources (ChEMBL, BindingDB, and BioLip2) and employes aggressive filtering from each of the datasets followed by molecules standardization, PAINS filtering and deduplication. The val/test sets are additionally binarized for binding event classification at a threshold of < 1 μM on Ki/Kd to have unified benchmark to compare models on. PLINDER is employed to split the proteins into training and testing sets. To enhance flexibility, the training set includes continuous values and their corresponding signs (=, >, <).
Note: PLUMBER states for Protein–Ligand Unseen Matching Benchmark for Evaluating Robustness
PLINDER
To develop generalizable sequence/structure-based models, we aim to test our model on unseen proteins. Standard techniques, such as time-split and random split, often result in test sets containing many very similar proteins, which limits the ability to measure generalizability. The recent benchmark, PLINDER, proposed a compound metric that accounts for different types of similarity on system level and splits datasets based on this metric. We decided to use their protein split assignment. While it is not perfect (as we lack ligand split information), it should yield more challenging splits compared to standard techniques.
Data description
val and test sets contain the following columns:
SMILES: standardized SMILES representation of a moleculesequence: amino acid sequence of a monomer target proteinuniprot_id: UniProt ID of that proteinsource: either "chembl", "bdb", or "biolip"split: always set to "test"is_active: a binary label indicating if the molecule has a Ki/Kd < 1 μM
train set does not have an is_active column. Instead, it contains different columns useful for training. ki, kd, ic50, and ec50 store activity values, while ki_sign, kd_sign, ic50_sign, and ec50_sign specify the corresponding relationship, such as =, <, or >. You can choose to use only Ki/Kd equality data, but alternative strategies can incorporate the other activity types as well.
Preprocessing
To ensure high data quality, we performed extensive preprocessing steps:
- Selected only monomer data
- Standardized SMILES with ChEMBL structure pipeline
- Cleaned SMILES
- Prioritized data from BindingDB
- Filtered out molecules with PAINS filter
- Binarized and deduplicated values with inconsistency check for val/test sets
Acknowledgements
Many thanks to the PLINDER authors for the groundbreaking work on the advanced molecular data splitting.