
Background
Molecular representations are crucial for understanding molecular structure, predicting properties, QSAR studies, toxicology and chemical modeling and other aspects in drug discovery tasks. Therefore, benchmarks for molecular representations are critical tools that drive progress in the field of computational chemistry and drug design. In recent years, many large models have been trained for learning molecular representation. The aim is to evaluate large pretrained models are capable of predicting various “easy-to-compute” molecular properties.
Benchmarking
The objective is to comprehend the proficiency of a model in predicting these 'easy' properties, gauging its effectiveness. Ideally, any pre-trained models should, at the very least, demonstrate good performance in those tasks before applying them to the downstream tasks.
Description of readout
The computed properties are molecular weight, fraction of sp3 carbon atoms (fsp3), number of rotatable bonds, topological polar surface area, computed logP, formal charge, number of charged atoms, refractivity and number of aromatic rings. These properties are widely used in molecule design and molecule prioritization.
Number of data points: train: 202184, test: 47816
Data resource
Reference: https://pubs.acs.org/doi/10.1021/acs.jcim.2c01253
Raw data: https://cartblanche22.docking.org
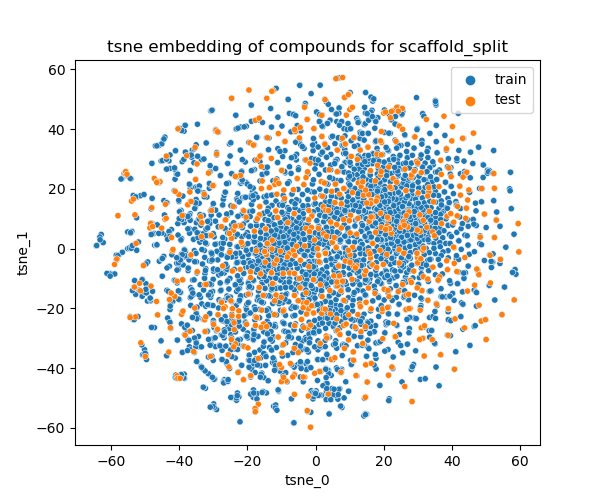
Train/test split
The objective is to comprehend the proficiency of a model in predicting these 'easy' properties. In order to select the predictive models which is able to generalize to new chemical space, a scaffold split is used to generate trian/test sets.
Distribution of the train/test in the chemical space
Related links
The full curation and creation process is documented here.
Related benchmarks